.svg)
.svg)

.svg)

ترجمه مقاله توقف مسیر اتصال پایانی غیر همولوگی (NHEJ)، توسط مهار کننده زیرواحد کاتالیزوری کیناز پروتئین وابسته به DNA - نشریه NATURE

عنوان فارسی
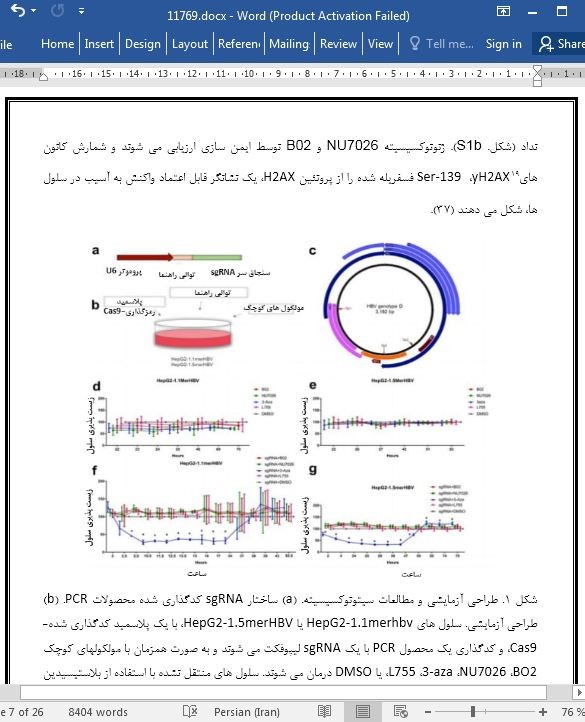
توقف مسیر اتصال پایانی غیر همولوگی (NHEJ)، توسط مهارکننده زیرواحد کاتالیزوری کیناز پروتئین وابسته به DNA (DNA-PKcs)، با نام NU7026 مانع از تخریب HBV cccDNA توسط کریسپر/کَس 9 (CRISPR/Cas9) می شود
عنوان انگلیسی
Suppressing the NHEJ pathway by DNA-PKcs inhibitor NU7026 prevents degradation of HBV cccDNA cleaved by CRISPR/Cas9
صفحات مقاله فارسی
26
صفحات مقاله انگلیسی
11
سال انتشار
2019
رفرنس
دارای رفرنس در داخل متن و انتهای مقاله
نشریه
NATURE
فرمت مقاله انگلیسی
pdf و ورد تایپ شده با قابلیت ویرایش
فرمت ترجمه مقاله
pdf و ورد تایپ شده با قابلیت ویرایش
فونت ترجمه مقاله
بی نازنین
سایز ترجمه مقاله
14
نوع مقاله
ISI
نوع ارائه مقاله
ژورنال
پایگاه
اسکوپوس
ایمپکت فاکتور(IF) مجله
4.130 در سال 2020
شاخص H_index مجله
213 در سال 2021
شاخص SJR مجله
1.240 در سال 2020
شناسه ISSN مجله
2045-2322
شاخص Q یا Quartile (چارک)
Q1 در سال 2020
کد محصول
11769
وضعیت ترجمه عناوین تصاویر
ترجمه شده است ✓
وضعیت ترجمه متون داخل تصاویر
ترجمه شده است ✓
وضعیت ترجمه منابع داخل متن
به صورت عدد درج شده است ✓
وضعیت فرمولها و محاسبات در فایل ترجمه
ندارد☓
ضمیمه
ندارد☓
بیس
است✓
مدل مفهومی
ندارد☓
پرسشنامه
ندارد☓
متغیر
ندارد☓
فرضیه
ندارد☓
رفرنس در ترجمه
در داخل متن و انتهای مقاله درج شده است
رشته و گرایشهای مرتبط با این مقاله
پزشکی و غدد و متابولیسم، ویروس شناسی پزشکی
مجله
گزارش های علمی - Scientific Reports
دانشگاه
موسسه تحقیقات مرکزی اپیدمیولوژی، هپاتیت ویروسی، فدراسیون روسیه
doi یا شناسه دیجیتال
https://doi.org/10.1038/s41598-019-38526-6
۰.۰
(هنوز امتیازی ثبت نشده است)