.svg)
.svg)

.svg)

ترجمه مقاله جهش های GABBR2 تعیین کننده فنوتیپ در سندرم رت و بیماری های مغزی صرعی - نشریه وایلی

عنوان فارسی
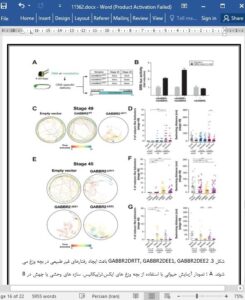
جهش های GABBR2 تعیین کننده فنوتیپ در سندرم رت و بیماری های مغزی صرعی
عنوان انگلیسی
GABBR2 Mutations Determine Phenotype in Rett Syndrome and Epileptic Encephalopathy
صفحات مقاله فارسی
22
صفحات مقاله انگلیسی
13
سال انتشار
2017
رفرنس
دارای رفرنس در داخل متن و انتهای مقاله
نشریه
وایلی - Wiley
فرمت مقاله انگلیسی
pdf و ورد تایپ شده با قابلیت ویرایش
فرمت ترجمه مقاله
pdf و ورد تایپ شده با قابلیت ویرایش
فونت ترجمه مقاله
بی نازنین
سایز ترجمه مقاله
14
نوع مقاله
ISI
نوع نگارش
مقاله پژوهشی (Research article)
نوع ارائه مقاله
ژورنال
پایگاه
اسکوپوس
ایمپکت فاکتور(IF) مجله
9.380 در سال 2019
شاخص H_index مجله
287 در سال 2020
شاخص SJR مجله
4.900 در سال 2019
شناسه ISSN مجله
1531-8249
شاخص Q یا Quartile (چارک)
Q1 در سال 2019
کد محصول
11562
وضعیت ترجمه عناوین تصاویر و جداول
ترجمه شده است ✓
وضعیت ترجمه متون داخل تصاویر و جداول
ترجمه نشده است ☓
وضعیت ترجمه منابع داخل متن
ترجمه نشده است ☓
وضعیت فرمولها و محاسبات در فایل ترجمه
ندارد
ضمیمه
ندارد
بیس
نیست ☓
مدل مفهومی
ندارد ☓
پرسشنامه
ندارد ☓
متغیر
ندارد ☓
رفرنس در ترجمه
در داخل متن و انتهای مقاله درج شده است
رشته و گرایش های مرتبط با این مقاله
پزشکی و مغز و اعصاب، ژنتیک پزشکی
مجله
سالانه مغز و اعصاب - ANNALS of Neurology
دانشگاه
گروه علوم پزشکی، کالج پزشکی دانشگاه ملی سئول، کره جنوبی
doi یا شناسه دیجیتال
https://doi.org/10.1002/ana.25032
۰.۰
(هنوز امتیازی ثبت نشده است)